MULTICONFIGURATIONAL QUANTUM CHEMISTRY ON GPUS
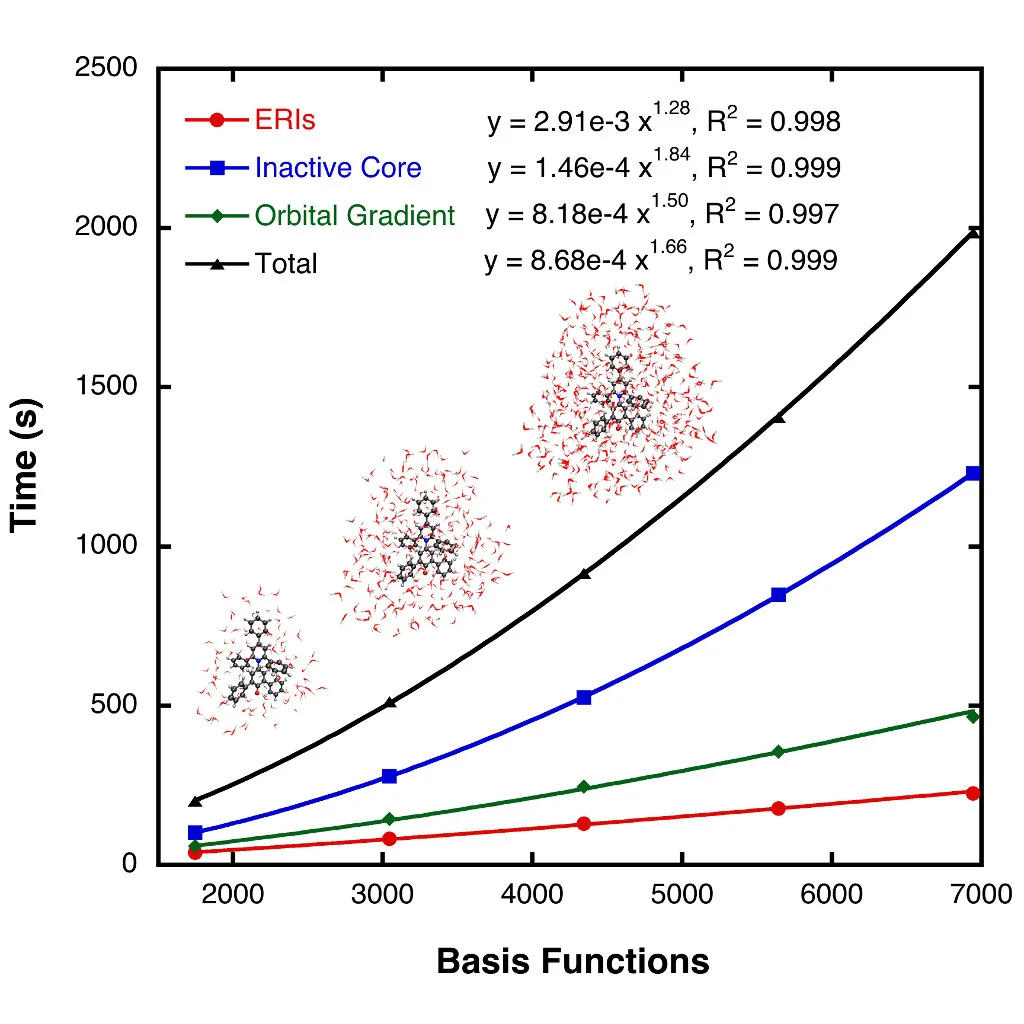
GPU Accelerated CASSCF
We have developed an algorithm for the CASSCF orbital optimization that uses sparsity in the atomic orbital (AO) basis set to extend the applicability of CASSCF to systems of unprecedented size. Our implementation of this algorithm uses graphical processing units (GPUs) and has allowed us to perform CASSCF computations on molecular systems containing more than one thousand atoms. Read the paper.
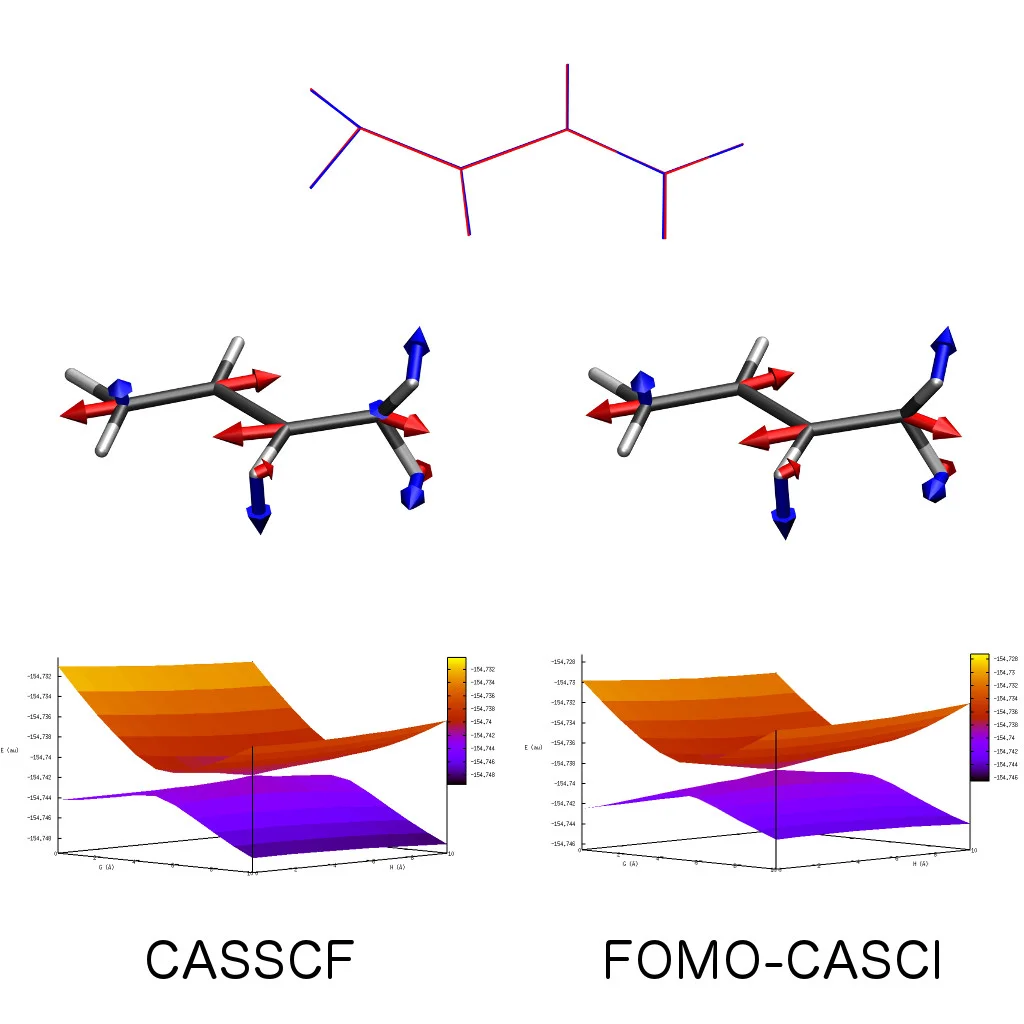
Efficient Approximations to CASSCF
The floating occupation molecular orbital-complete active space configuration interaction (FOMO-CASCI) method is a promising alternative to the state-averaged complete active space self-consistent field (SA-CASSCF) method. We have formulated the analytic first derivative of FOMO-CASCI in a manner that is well-suited for a highly efficient implementation using graphical processing units (GPUs). This implementation of FOMO-CASCI is of similar computational expense to configuration interaction singles (CIS) or time-dependent density functional theory (TDDFT). In contrast to CIS and TDDFT, FOMO-CASCI can describe electronic wavefunctions with significant multireference character. Read the paper.